
Research – Open Access
Min Li, Wenye Zhu, Ummair Saeed, Shibo Sun, Yan Fang, Chu Wang & Zhuang Luo
BMC Pulmonary Medicine volume 22, Article number: 29 (2022)
Abstract
Background
Asthma is a heterogeneous disease and different phenotypes based on clinical parameters have been identified. However, the molecular subgroups of asthma defined by gene expression profiles of induced sputum have been rarely reported.
Methods
We re-analyzed the asthma transcriptional profiles of the dataset of GSE45111. A deep bioinformatics analysis was performed. We classified 47 asthma cases into different subgroups using unsupervised consensus clustering analysis. Clinical features of the subgroups were characterized, and their biological function and immune status were analyzed using Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) and single sample Gene Set Enrichment Analysis (ssGSEA). Weighted gene co-expression network analysis (WGCNA) and protein–protein interaction (PPI) network were performed to identify key gene modules and hub genes.
Results
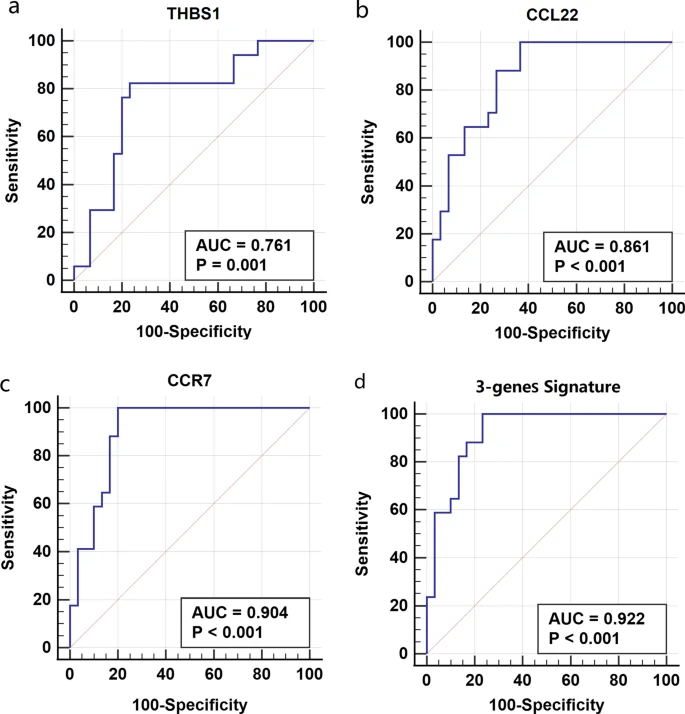
Unsupervised consensus clustering of gene expression profiles in asthma identified two distinct subgroups (Cluster I/II), which were significantly associated with eosinophilic asthma (EA) and paucigranulocytic asthma (PGA). The differentially expressed genes (DEGs) between the two subgroups were primarily enriched in immune response regulation and signal transduction. The ssGSEA suggested the different immune infiltration and function scores between the two clusters. The WGCNA and PPI analysis identified three hub genes: THBS1, CCL22 and CCR7. ROC analysis further suggested that the three hub genes had a good ability to differentiate the Cluster I from the Cluster II.
Conclusions
Based on the gene expression profiles of the induced sputum, we identified two asthma subgroups, which revealed different clinical characteristics, gene expression patterns, biological functions and immune status. The transcriptional classification confirms the molecular heterogeneity of asthma and provides a framework for more in-depth research on the mechanisms of asthma.